






肿瘤细胞一大特征为细胞能量代谢的重编程,而这种重编程主要依赖于基因组的不稳定性和易变性赋予癌细胞基因改变,从而推动肿瘤的发展。肿瘤细胞基因组的高通量DNA测序分析揭示了某些人类肿瘤的体细胞突变,这些突变预示了通常由激活的生长因子受体触发的信号通路的持续激活(如FLT3)。
“代谢重编程”和“代谢重组”这两个术语的出现与使用,可以更好的理解肿瘤细胞中的代谢改变。从定义的角度来看,“代谢重编程”代表了癌细胞的“软件变化”,描述了这种增殖细胞通常由于生长因子的诱导,但会被致癌信号阻断;而“代谢重组”则代表了癌细胞的“硬件改变”,并描述了由于癌基因突变体的新功能而引起的代谢改变,这在正常细胞中是没有发现的。胶质瘤和急性髓系白血病(AML)中异柠檬酸脱氢酶(IDH)1和2突变的鉴定代表了一个重新整合的过程,因为这些突变赋予IDH1/2新功能,产生肿瘤代谢物2-羟基谷氨酸(2-HG)来调节癌症表观遗传学,而这在携带野生型(WT)IDH1/2的正常细胞中是不存在的。

这项研究由埃默里大学医学院陈静团队完成,并发表在《Cancer Discovery》上(2019 IF: 29.497),题目为“Mutant and Wild-Type Isocitrate Dehydrogenase 1 Share Enhancing Mechanisms Involving Distinct Tyrosine Kinase Cascades in Cancer”
陈静教授团队证明了致癌的酪氨酸激酶(TKs)与野生型和突变型IDH1的激活之间的内在联系,这涉及相关癌症中不同的酪氨酸激酶级联。特别强调的是,这些结果为突变型IDH1阳性AML提供了一种有前途的临床治疗方法,即为FLT3和突变型IDH1抑制剂的联合应用提供了一个新的理论基础。
FLT3和IDH1的内在关系
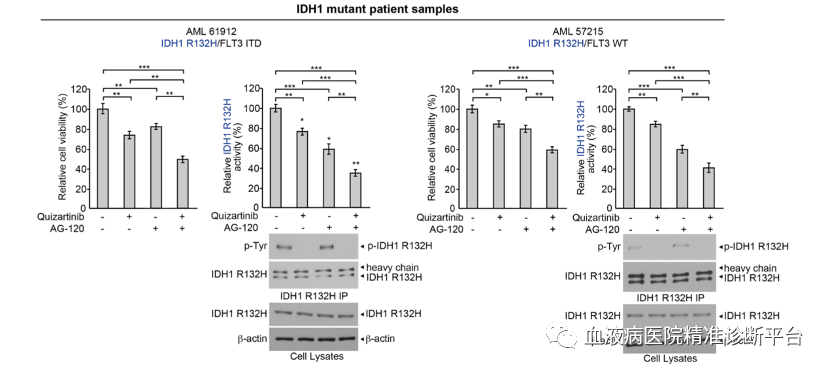
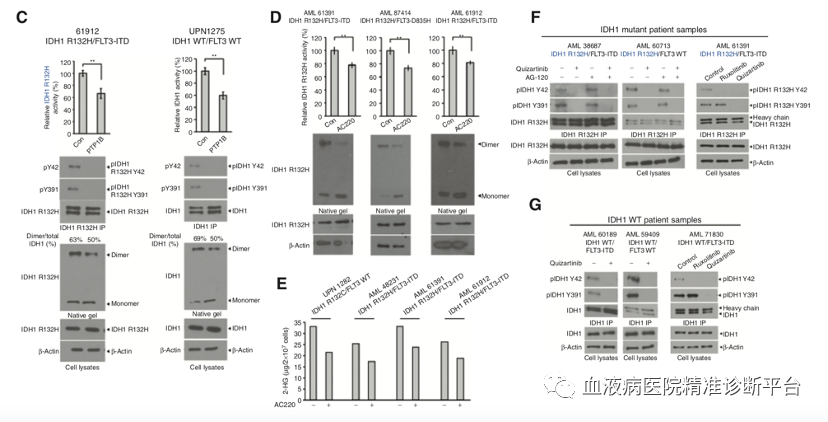
研究人员首先利用FLT3激酶抑制剂奎扎替尼与IDH1 R132位突变抑制剂AG-120共处理人源AML细胞(分别携带IDH1 R132H/FLT3 ITD或IDH1 R132H/FLT3 WT),发现奎扎替尼或AG-120单独处理AML细胞(分别携带IDH1 R132H/FLT3 ITD或IDH1 R132H/FLT3 WT)均可不同程度抑制细胞活力,而二者共同使用会进一步降低细胞活力,且这种抑制趋势同样见于IDH1 R132酶活性。奎扎替尼处理IDH1 WT/FLT3 ITD的AML细胞同样会降低细胞活力及IDH1 WT的酶活性。这种不依赖于激酶突变状态的关系很可能是由于FLT3依赖的IDH1酪氨酸位点磷酸化。
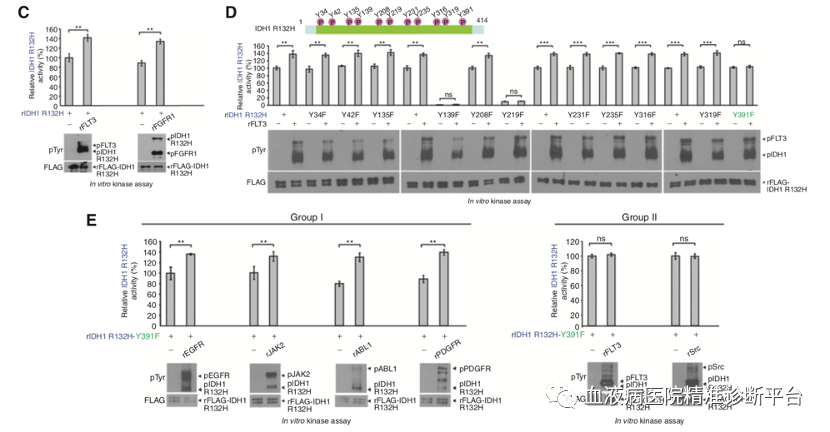
不同的酪氨酸激酶磷酸化IDH1不同的酪氨酸位点
利用体外重组表达纯化的IDH1 R132H(rIDH1 R132H)突变蛋白,与各类具有重组活性的酪氨酸激酶蛋白(包括rFLT3, rFGFR1, rSrc, rPDGFR, rEGFR, rMET, rKIT和rJAK2)孵育,发现所有具有重组活性的酪氨酸激酶蛋白均可直接且显著的磷酸化纯化的rIDH1 R132H突变体,导致突变体IDH1活性增强。研究者进一步寻找并发现IDH1 Y139和Y219突变会内在地废除IDH1突变体活性,而IDH1 Y391F会消除FLT3活化IDH1突变体的能力。进一步研究发现这些酪氨酸激酶可以分成两类,其中1类包括EGFR, JAK2, ABL1, PDGFR, MET, FGFR1和 FGFR3只通过Y42位磷酸化激活IDH1 ;而2类包括FLT3和Src,是通过Y391位磷酸化激活IDH1。这些数据表明,这两类TKs均可通过IDH1的Y42或Y391位的磷酸化方式活化IDH1 WT或R132H突变体,这些结果表明WT和突变型IDH1可能具有相同的酪氨酸磷酸化调控机制。
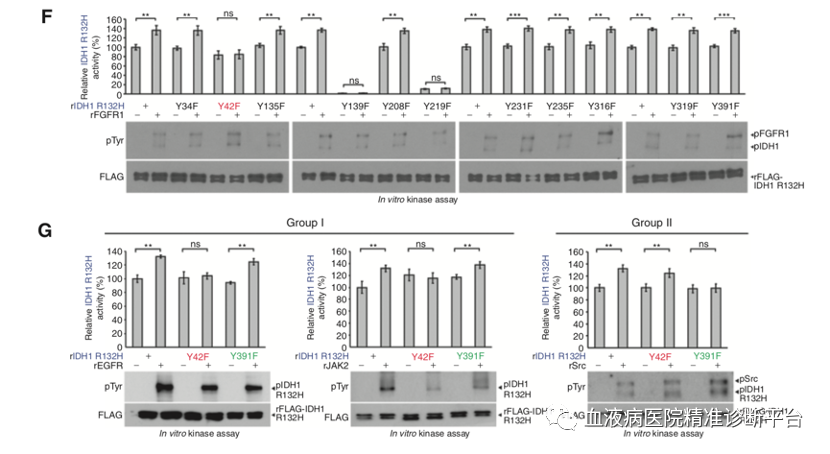
Y42位磷酸化促进IDH1二聚化和随之的底物结合
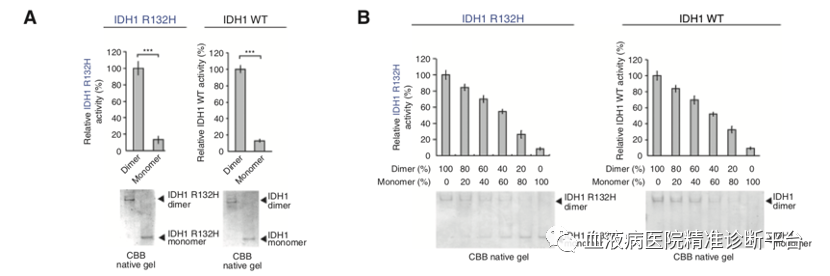
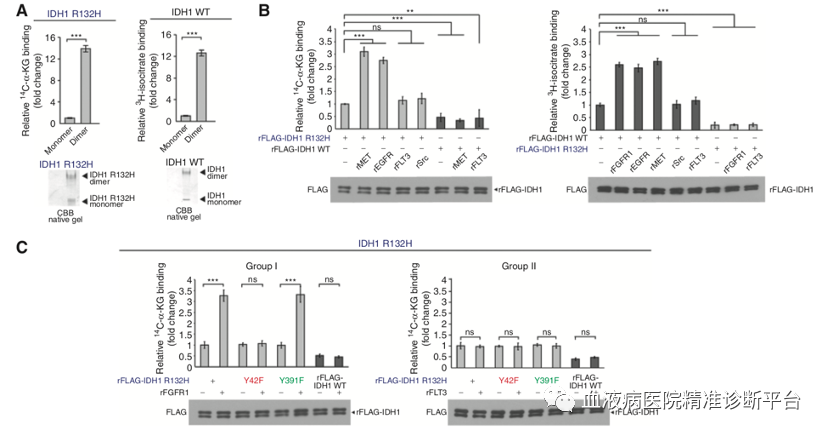
接着研究团队想弄清Y42和Y391位磷酸化分别扮演何种角色。纯化的 IDH1 R132H 突变型或野生型蛋白的二聚体和单体分别代表IDH1的活性和非活性形式,并且IDH1的活性随着二聚体与单体比例的提高而升高。进一步研究表明,1类的TKs(而不是2类),可以显著上调IDH1 R132H 突变体与野生型的二聚体,而1类的TKs是通过Y42位磷酸化调控IDH1活性,表明Y42位磷酸化可能调控IDH1的二聚体形成。进而研究者通过一系列生化实验进一步证明了这个观点。
在确定了Y42位磷酸化增强二聚体形成促进IDH1激活的机制后,又发现IDH1二聚体的IDH1突变体或野生型对14C标记的α-酮戊二酸(14C-αHG)或3H标记的柠檬酸盐具有高亲和力,表明IDH1二聚体的形成促进了与底物的结合。有趣的是,包含rMET和rEGFR的1类TKs会促进14C-αHG结合IDH1突变体,3H-柠檬酸盐结合IDH1野生型,而包含FLT3的2类TKs不影响这种结合。
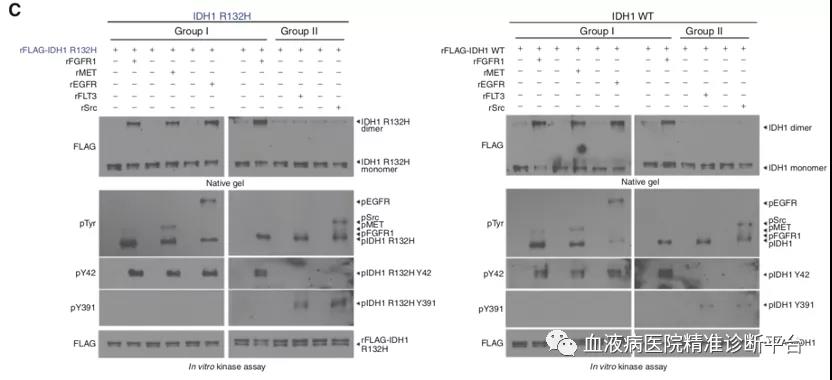
Y391位磷酸化促进NADP+结合IDH1
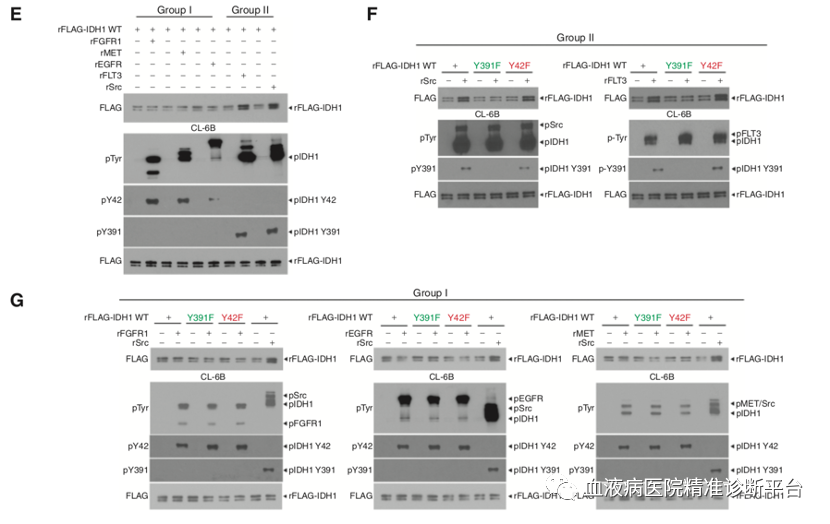
IDH1的各种结构模型预测都显示了Y391位靠近NADP+的结合位点。为弄清这一猜想,研究人员利用了一种被称为CL-6B琼脂糖珠的材料,CL-6B可以模拟NADP+,是以NADP+为底物的多种脱氢酶的假亲和配体。结果表明,包含rFLT3和rSrc的2类TKs(而不是1类TKs)会导致与CL-6B琼脂糖珠结合的IDH1蛋白量显著增加,显示IDH1与NADP+的结合增强,表明2类TKs诱发的Y391位磷酸化可能参与了辅因子NADP+与IDH1的结合。进一步研究显示,只有Src和FLT3能够促进IDH1野生型和Y42F突变型结合CL-6B琼脂糖珠,而不是Y391F突变型,表明2类TKs诱发的Y391为磷酸化促进NADP+结合IDH1。
Y42磷酸化只发生在IDH1单体中,而Y391磷酸化可发生在单体和二聚体IDH1中
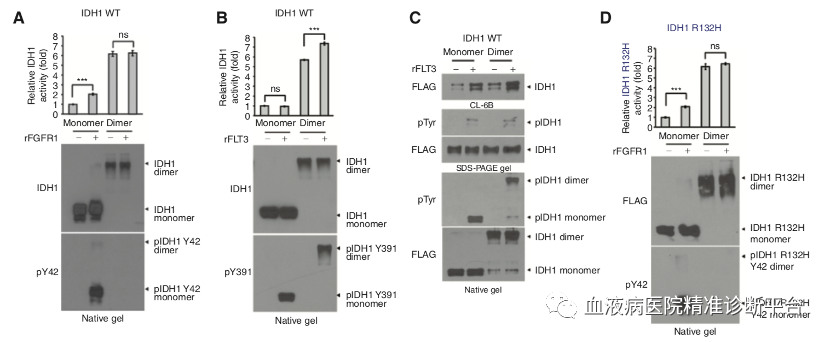
FGFR1诱导的Y42位磷酸化只发生于纯化的单体IDH1野生型或突变型中,进而促进IDH1单体向二聚体转化,增强酶活性;而FGFR1不能磷酸化纯化的二聚体IDH1野生型蛋白Y42位,也不会改变二聚体含量和酶活性。相反,FLT3诱导的Y391位磷酸化在纯化的单体或二聚体IDH1蛋白中均可发生,单体IDH1的Y391位磷酸化不会促进二聚体的形成,但会增强酶活性,这是因为增强了辅因子NADP+的结合。
事实上,单体IDH1野生型或突变型蛋白会自发不同程度地时间依赖性的形成二聚体,这也解释了非磷酸化的单体IDH1野生型或突变型蛋白也会有本底水平的酶活性。而二聚体IDH1野生型或突变型蛋白也会自发不同程度地时间依赖性的转化成单体,但rFGFR1会抑制这种转化。这些数据共同表明,Y42磷酸化发生在IDH1单体中,促进二聚体形成,一旦二聚体形成,Y42磷酸化稳固二聚体状态。
FLT3 WT或ITD突变型通过直接磷酸化Y391和间接磷酸化Y42来增强AML细胞中IDH1 WT或突变型的活性
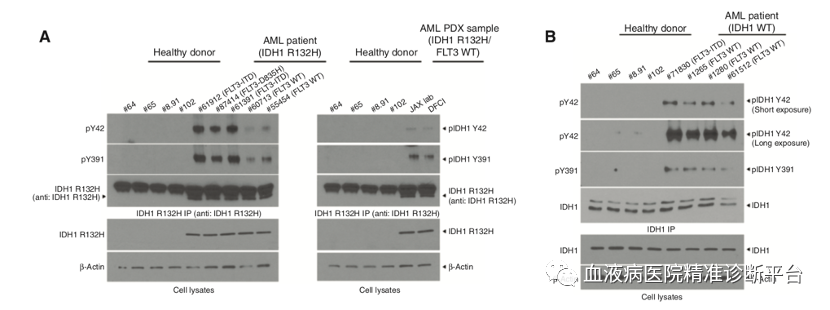
尽管FLT3突变状态不同,但从IDH1 R132H突变的AML患者的原代白血病细胞中检测到Y42和Y391位的磷酸化;与健康者的外周血细胞对照相比,具有代表性IDH1-WT的AML患者的原代白血病细胞中的Y42和Y391位磷酸化水平显著上调。蛋白酪氨酸磷酸酶处理IDH1 R132H突变或WT的AML患者的原代白血病细胞会抑制IDH1活性,降低Y42和Y391磷酸化水平,下调IDH1蛋白二聚体总量。此外FLT3抑制剂奎扎替尼处理IDH1 R132H/FLT3 ITD突变的AML患者的原代白血病细胞会抑制IDH1活性,降低Y42和Y391磷酸化水平,下调IDH1蛋白二聚体,减少2-羟基谷氨酸产生,而JAK2抑制剂只能下调Y42磷酸化水平。
总结
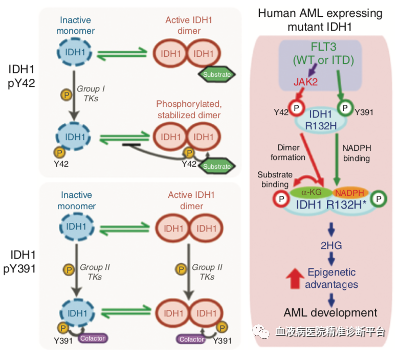
FLT3(不论是野生型或突变型)可以间接通过JAK2磷酸化IDH1的 Y42位,也可以直接磷酸化IDH1 的Y391位,二个磷酸化位点分别通过调节IDH1二聚化及与NADPH底物的结合能力,促进了IDH1突变型肿瘤细胞产生2-羟基谷氨酸(2-HG),而由野生型IDH1催化产生的α-酮戊二酸(α-KG)的减少,从而影响了α-KG为配体的JHD1和TET2基因,促进AML的发生与发展。本研究为携带酪氨酸激酶或IDH1基因突变的AML患者联合用药提供了理论指导。
参考文献:
1. Hanahan D, Weinberg R A. Hallmarks of Cancer: The Next Generation[J]. Cell, 2011, 144(5): 646-674.
2. Cairns R A, Mak T W. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities[J]. Cancer Discovery, 2013, 3(7): 730-741.
撰稿:黄丙庆
编辑:曲晓宁
审核:王慧君 孙琦
 津公网安备12010102000385号
津公网安备12010102000385号